New Tool Allows Unprecedented Modeling of Magnetic Nanoparticles
For Immediate Release
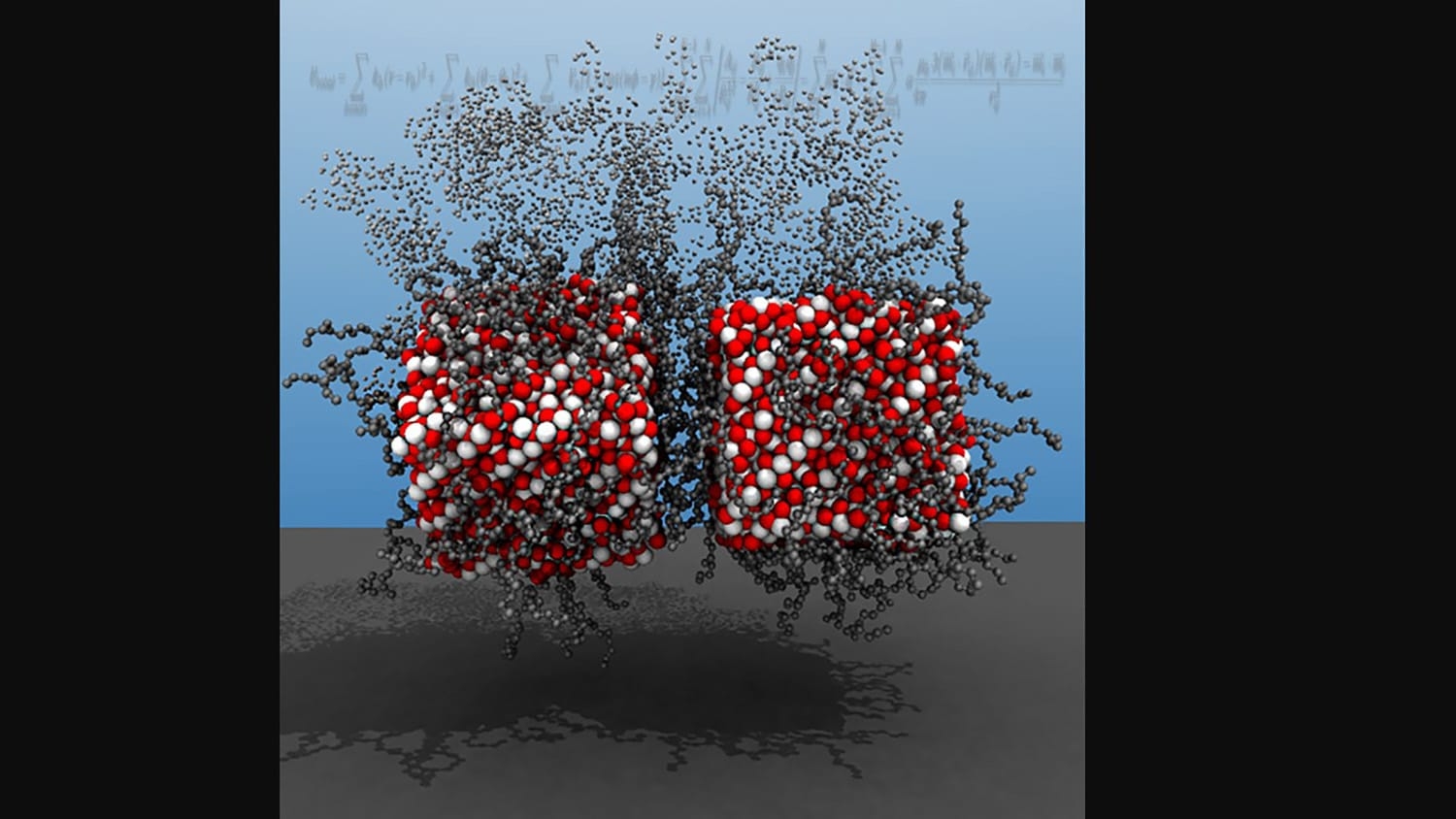
Researchers at North Carolina State University have developed a new computational tool that allows users to conduct simulations of multi-functional magnetic nanoparticles in unprecedented detail. The advance paves the way for new work aimed at developing magnetic nanoparticles for use in applications from drug delivery to sensor technologies.
“Self-assembling magnetic nanoparticles, or MNPs, have a lot of desirable properties,” says Yaroslava Yingling, corresponding author of a paper on the work and a Distinguished Professor of Materials Science and Engineering at NC State. “But it has been challenging to study them, because computational models have struggled to account for all of the forces that can influence these materials. MNPs are subject to a complicated interplay between external magnetic fields and van der Waals, electrostatic, dipolar, steric, and hydrodynamic interactions.”
Many applications of MNPs require an understanding of how the nanoparticles will behave in complex environments, such as using MNPs to deliver a specific protein or drug molecule to a targeted cancer affected cell using external magnetic fields. In these cases, it is important to be able to accurately model how MNPs will respond to different chemical environments. Previous computational modeling techniques that looked at MNPs were unable to account for all of the chemical interactions MNPs experience in a given colloidal or biological environment, instead focusing primarily on physical interactions.
“Those chemical interactions can play an important role in the functionality of the MNPs and how they respond to their environment,” says Akhlak Ul-Mahmood, first author of the paper and a Ph.D. student at NC State. “And detailed computational modeling of MNPs is important because models offer an efficient path for us to engineer MNPs for specific applications.
“That’s why we’ve developed a method that accounts for all of these interactions, and created open-source software that the materials science community can use to implement it.”
“We’re optimistic that this will facilitate significant new research on multi-functional MNPs,” Yingling says.
To demonstrate the accuracy of the new tool, the researchers focused on oleic acid ligand-functionalized magnetite nanoparticles, which have already been studied and are well-understood.
“We found that our tool’s predictions of the behavior and properties of these nanoparticles was consistent with what we know about these nanoparticles based on experimental observation,” Mahmood says.
What’s more, the model also offered new insights into the behavior of these MNPs during self-assembly.
“We think the demonstration not only shows that our tool works, but highlights the additional value that it can provide in terms of helping us understand how best to engineer these materials in order to leverage their properties,” Yingling says.
The paper, “All-Atom Simulation Method for Zeeman Alignment and Dipolar Assembly of Magnetic Nanoparticles,” is published in the Journal of Chemical Theory and Computation. The work was done in collaboration with the experimental group of Joe Tracy, a professor of materials science and engineering at NC State, and with support from the National Science Foundation, under grant number CMMI-1763025.
-shipman-
Note to Editors: The study abstract follows.
“All-Atom Simulation Method for Zeeman Alignment and Dipolar Assembly of Magnetic Nanoparticles”
Authors: Akhlak U. Mahmood and Yaroslava G.Yingling, North Carolina State University
Published: March 10, Journal of Chemical Theory and Computation
DOI: 10.1021/acs.jctc.1c01253
Abstract: Magnetic nanoparticles (MNPs) can organize into novel structures in solutions with excellent order and unique geometries. However, studies of the self-assembly of smaller MNPs are challenging due to a complicated interplay between external magnetic fields and van der Waals, electrostatic, dipolar, steric, and hydrodynamic interactions. Here, we present a novel all-atom molecular dynamics (AMD) simulation method to enable detailed studies of the dynamics, self-assembly, structure and properties of MNPs as a function of core sizes and shapes, ligand chemistry, solvent properties and external field. We demonstrate the use and effectiveness of the model by simulating the self-assembly of oleic acid ligand-functionalized magnetite (Fe3O4) nanoparticles, with spherical and cubic shapes, into rings, lines, chains, and clusters under a uniform external magnetic field. We found that the long-range electrostatic interactions can favor the formation of a chain over a ring, the ligands promote MNP cluster growth, and the solvent can reduce the rotational diffusion of the MNPs. The algorithm has been parallelized to take advantage of multiple processors of a modern computer and can be used as a plugin for the popular simulation software LAMMPS to study the behavior of small magnetic nanoparticles and gain insights into the physics and chemistry of different magnetic assembly processes with atomistic details.
This post was originally published in NC State News.


