
Featured Projects
Sequencing Florida panther genome
The NC State University Genomic Sciences Laboratory (GSL) partnered with High School students in Milton, Florida to sequence the genome of the rare and endangered Florida panther (Puma concolor coryi), an isolated population of the North American cougar or mountain lion (Puma concolor). Currently, only 120-230 Florida panthers are thought to exist, and are located largely south of Lake Okeechobee in the natural habitat of the Florida Everglades and Big Cypress Region. These animals ranged historically throughout Florida and other parts of the southeastern United States. The Florida panther is currently under the protection of the Endangered Species Act1.

The Florida panther is the mascot of Milton High School, located in the small town of Milton, Florida in Santa Rosa County of the Florida Panhandle. Sam Rezell, a Milton High School science teacher, partnered with a former Milton High School alumnus, Mike Kreitzinger, who is currently a regional sales director for Illumina, Inc. Illumina is a San Diego-based genomics company whose mission is the unlock the power of the genome to improve human health. Rezell and Kreitzinger partnered with Dr. Dave Onorato of the Florida Fish & Wildlife Conservation Commission (FWC) to host a three-day sequencing workshop at Milton High School in which students learned about the significance and applications of DNA sequencing, as well as gained hands-on experience with pipetting and DNA sequencing procedures. As a final component of the workshop, Illumina lent a MiSeq System so students could sequence the actual genome of a real Florida panther, identified as FP20 by FWC panther biologists2.
Panther FP20 was initially captured and radio collared by FWC in March of 1987. He was subsequently injured in a vehicle collision in June of 1987. FP20 was treated and rehabilitated at Miami Metro Zoo (now Zoo Miami) until August of 1987, when he was released back into the wild (Fig. 1). FP20 lived for another year, and died in the wild in August of 1988, from issues associated with a heart condition (atrial septal defect) found to be prevalent in Florida panthers during that era due to inbreeding depression3. A sample of DNA was taken during his convalescence at the Miami Metro Zoo, and this was used for the Florida panther genome sequencing efforts. In addition to
providing invaluable research experience for Milton High School students, enabling them to perform cutting-edge research on their school mascot, the sequenced Florida panther genome has enormous potential to enhance conservation efforts for this species. In a collaborative effort, the GSL facility at NC State University has agreed to provide additional sequencing data for this project, in order to generate a high-quality reference genome that can be used to better understand the population structure of Florida panthers in the wild, develop better forensic testing methods to protect the status of this endangered animal, and to foster a greater understanding of animal diseases in the wild.

Genomic Resources For Sweet Potato Improvement
Yencho Lab
Sweetpotato is the seventh most important food crop due to its high adaptability to tropical, subtropical and temperate climates. Its low glycemic index, high fiber content and nutraceutical content is making it popular in the chip and French fry industry. It is a critical food and health security crop in many developing countries, especially in sub-Saharan Africa. Its wide adoptability is still limited by cultural preference for specific culinary and agronomic traits and challenges due to its polyploid nature and resulting complex trait inheritance.

With funding support from the Bill and Melinda Gates Foundation Genomic Tools for Sweetpotato (GT4SP) Improvement Project and Illumina, Inc., Greater Good Initiative we are developing genomic resources for this autoallopolyploid. These include: (i) reference genome assemblies of two of sweetpotato’s wild diploid progenitors; and (ii) an optimized sequencing-based genotyping protocol and analysis pipeline that provides accurate dosage calls, minimal missing data and high-fidelity SNPs. Ongoing efforts to generate additional genomic resources include: (i) reference genome assembly of tropical- and temperate-adapted hexaploid sweetpotato genomes; and (ii) developing a SNP panel (for a SNP array or targeted sequencing-based genotyping) with minimal ascertainment bias. These will be derived from our ongoing effort to provide high-density DNA markers for the germplasm collection representing the global diversity of the crop.
Beyond the generation over 20 Tb of sequencing data, we hope to provide insights into fundamental biological questions and show how these resources can be routinely deployed in breeding programs. Currently, some of our studies include understanding the genetic basis for rapid climatic adaptation of crops and the genomic features that are perturbed during this process. Using dosage information of polyploid genotypes, we are constructing linkage maps and performing QTL and genome-wide association analysis for several important traits. Results from these studies will encourage use of marker-assisted and genomic selection in sweetpotato breeding programs worldwide.

Regions that control the plant defense response
Dr. Yijian He, Dr. Shannon Sermons, and Dr. Peter Balint-Kurti

Cell death plays a critical role in the plant immune system. Plants use the hypersensitive response (HR), a form of programed cell death in which rapid cell death occurs at the point of attempted pathogen penetration, to restrict the spread of infection. However, if not controlled, HR can lead to unnecessary tissue damage. Hence proper regulation of HR is critical. To study the regulation of HR in maize, we are interested in identifying expression quantitative trait loci (eQTLs), regions of the genome that regulate the transcriptional responses to the HR. The Rp1D21 gene can cause spontaneous HR in maize plants without any pathogen infection. We performed RNA sequencing using the Illumina Hiseq to detect the gene expression changes associated with HR caused by Rp1D21 in 100 lines from a genetic mapping population. The expression change of each individual gene will be considered to be a separate trait. eQTLs will be identified that are associated with changes in the expression of each gene. We are particularly interested in finding “eQTL hotspots” i.e. regions of the genome that are associated with changes in expression of multiple genes. eQTLs hotspots identified in this study will be compared to the regions we mapped previously that are associated with visible changes in the HR phenotype caused by Rp1D21 and with others that are associated with disease resistance to try to determine if there are genetic connections between these traits.
Viral Evolution in Action

Our NSF – PIRE grant focuses on the plasticity of the viral genomes. Geminiviruses are ssDNA viruses that replicate in nuclei of infected cells and are transmitted by insects. They are devastating in tropical and subtropical areas. We are interested in identifying bottlenecks for virus evolution in a cassava-geminivirus-whitefly pathosystem. We will inoculate the viruses onto cassava and then track the changes in the viral genome through time. We will examine viral diversity during transmission by insects and vegetative propagation in serial studies. We will be collecting DNA from cassava and whiteflies at each passage, amplify the viral genomes by rolling circle amplification, and sequence the genomes using the Illumina next generation platform. Comparison of the results will provide insight into the drivers of virus evolution and help prioritize efforts for control of these devastating pathogens.
Immune Haplotypes in Zebrafish
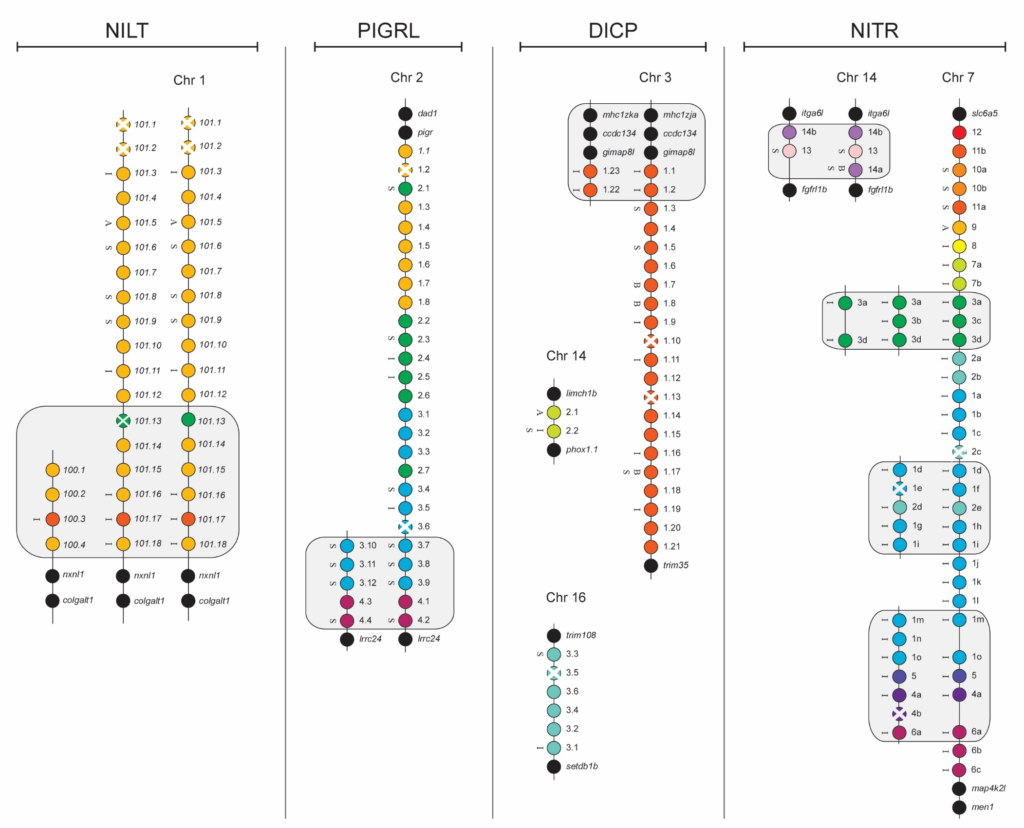
The zebrafish has proven to be an extremely amenable model system for genetic and developmental studies and more recently, the modeling of human disease. The availability of a high quality reference genome has accelerated these studies. However, resequencing of zebrafish genomic and transcriptomic loci has revealed the wide extent of not only SNPs but also gene content haplotypes within multigene families; individual zebrafish encode variable numbers of related genes due to frequent gene gain and loss events. Families of immune-related genes, including NITRs, DICPs, PIGRLs and NILTs, that are clustered in the genome exhibit dramatic gene content haplotypes. Whole Transcriptome sequencing using NGS platforms are helpful to resolve gene content haplotypes as prior knowledge of the sequence is unnecessary. De novo transcriptome assembly of the deep coverage obtained through RNAseq by NCSU’s GSL has allowed us to discover many novel immune-related genes absent from the latest versions of the zebrafish genome assembly. Currently, multiple projects in the Yoder lab (CVM) are underway to determine (i) the extent of gene content haplotypes in multiple genetic backgrounds of zebrafish and (ii) how gene content haplotypes observed in multigene families of innate immune receptors influence the individual response to immune challenge.
A Novel Application for an Ecological Pest
Invasive house mice on the Farallon Islands off the Coast of California are a model for invasive rodent populations on islands, which can exhibit unique ecological, genetic and physiological characteristics. Dr. John Godwin’s laboratory discovered that wild mice on these islands exhibit behavioral differences compared to the widely-used C57/BL6 laboratory strain in open field tests, particularly with regard to anxiety-related behaviors. Building on prior work from this group, that demonstrated distinct neurotranscriptomic profiles between shy and bold lines of zebrafish, the laboratory is currently working to characterize the neurotranscriptome patterns of Farallons mice in relation to those of C57/BL6s. These studies aim to elucidate genetics and expression profiles associated with anxiety in mice, which may lead to an improved mammalian model for the study of human anxiety disorders. In addition, by comparing the expression profiles of these mice with those from the zebrafish experiments, highly conserved gene expression patterns associated with anxiety may be identified, which could provide insights regarding relevant genes, neural pathways and mechanisms.
New Caviar Inspired Ecologically

North Carolina caviar, while delicious, is valued at 1/10th the price of some Russian caviars. In part, this may be because consumers desire the rarity of Russian caviar. But it is also possible that certain subtle flavors in Russian caviar simply make it more delicious. Can we make versions of NC caviar which are more like those of the Russians? Benjamin Reading, Robert Dunn, Scott Salger, and Lauren Nichols at the NC State Department of Applied Ecology have teamed together to characterize the microbes, tiny organisms living in caviar brine, using metagenomic DNA sequencing. While some microbes produce off flavors, our goal is to discover whether some microbes actually help to produce desirable flavors in caviar. If we find them, we can then add these tasty microbes to the brine of North Carolina caviar to make the flavors both more predictable and “rich”. We have already identified one potential microbe, which might be setting the highest quality caviar apart from all the rest. While we find this organism in all of the caviars that we have studied, it seems more common in the most expensive caviars. Now, we will attempt to study and grow this fascinating organism.
Transcriptomics in Loblolly pine

Loblolly pine breeding programs are faced with an interesting dilemma which includes long generation time (~25 years) and large scale progeny testing required for the advancement of genetically superior individuals. Currently, selections are made on the basis of family mean phenotype, where phenotypically superior individuals are selected from top-performing families, and progeny tested over a period of 6 years to screen for those trees that have the best breeding values. However, there is little confidence that phenotypically superior selections from a progeny test will carry forward traits intended from the family, because many traits of interest to breeders have low individual-tree heritability. To address this, we are exploring the relationship of transcriptomic data between 3-month old seedlings and stand age breeding values. RNA expression results from families with known breeding values will be used to create covariance matrices reflecting shared genetic variation in coding sequences and gene regulatory networks. The goal of this research is to identify underlying regulatory patterns which may correlate with breeding values and thereby help to reduce the time it takes to make forward selections. This work is being conducted by graduate student Adam Festa in the laboratory of Dr. Ross Whetten.
Arabidopsis Stem Cells
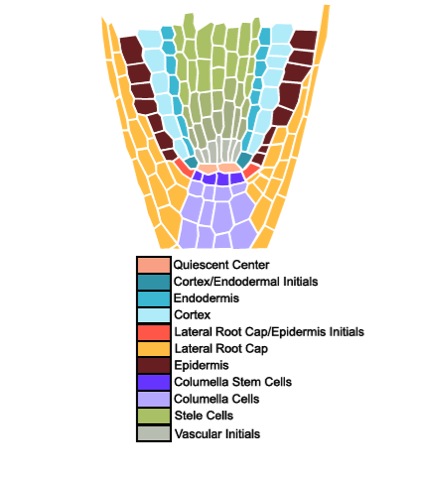
Stem cells divide to form all of the differentiated cell types in a multicellular organism. In the plant model organism Arabidopsis thaliana, the different stem cell populations surround a set of mitotically inactive cells called the quiescent center. Fluorescence activated cell sorting of the different stem cell populations in the quiescent center, coupled with cell-type specific transcriptomic profiling, are used to determine which genes function to maintain the stem cell populations in their undifferentiated form. Additionally, these same techniques are used in different mutant backgrounds to determine which genes function as differentiation factors for the different stem cell populations. This work is being conducted by graduate students Adam Fisher and Natalie Clark, postdoctoral scholar Dr. M. Angels de Luis Balaguer, and Dr. Ross Sozzani.
White Perch Proteomics
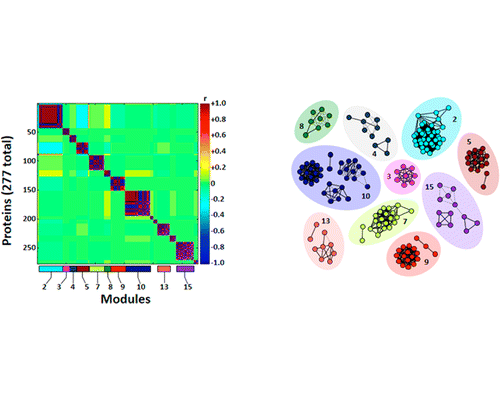
Transcriptome sequence assemblies enable Justin Schilling (Graduate Student, Department of Applied Ecology), Benjamin Reading (Department of Applied Ecology), and David Muddiman (Department of Chemistry) to perform semi-quantitative tandem mass spectrometry proteomics on a non-model species, the white perch. A compartment proteomics strategy was used to quantify proteins in cytosolic and membrane cell fractions of the ovary and protein expression patterns were used to train machine-learning models called support vector machines. These models were then able to accurately classify samples as either membrane or cytosolic fraction. The study opens up the possibilities of being able to identify unknown samples of tissue or cell fractions using proteomics. The research article was published in the Journal of Proteome Research (http://pubs.acs.org/doi/abs/10.1021/pr401067g).
The Red Flour Beetle
Dr. Nathaniel Grubbs (Lorenzen lab) is investigating novel genetic elements in the red flour beetle

The red flour beetle, Tribolium castaneum, possesses several maternal-effect, selfish genetic elements, all called Medea. Offspring of a heterozygous Medea mother must inherit at least one copy of the gene to survive, but distinct Medea elements cannot rescue each other. It is hypothesized that the different Medea elements act on the same biological pathway, but this pathway has not been elucidated, and only one element, M1, has been sequenced. We are sequencing BACs from the M4 genomic region using PacBio, in the hope that comparisons with M1 will shed light on Medea’s mode of action. M4 is located in a highly-repetive region, therefore PacBio (a long-read format) was selected.
Tobacco budworm

The Gould Lab is researching how insect genomes evolve in response to transgenic crops. The group focuses on two Lepidopteran pests: Heliothis virescens and Helicoverpa zea. By applying GBS (genotyping-by-sequencing) to specimens collected over 2 decades, they are working to identify which allele frequencies have changed over time in response to expansion of Bt crops in the landscape. These molecular signatures of response to selection may provide important insight into which genes are associated with field-evolved Bt resistance, and improve resistance management tactics in the future.